Ionis Neurology: Pipeline
The content on this page is intended for use by US healthcare professionals only.
Ionis continues to build upon its pioneering platform and foundational knowledge to develop medicines that can alter disease trajectory.1-4
Transforming Genomic Insights Into RNA-Targeted Innovation
Ionis’ RNA-targeted technology translates genomics insights into potential transformative medicines.5
Once a causative gene is identified, researchers can use this knowledge to develop antisense oligonucleotides (ASOs) that target the associated mRNA and modulate protein production.4,6,7 Ionis has used knowledge from the genomics revolution to develop FDA-approved medicines for the treatment of spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS), and polyneuropathy in patients with hereditary transthyretin (hATTR) amyloidosis.7-13
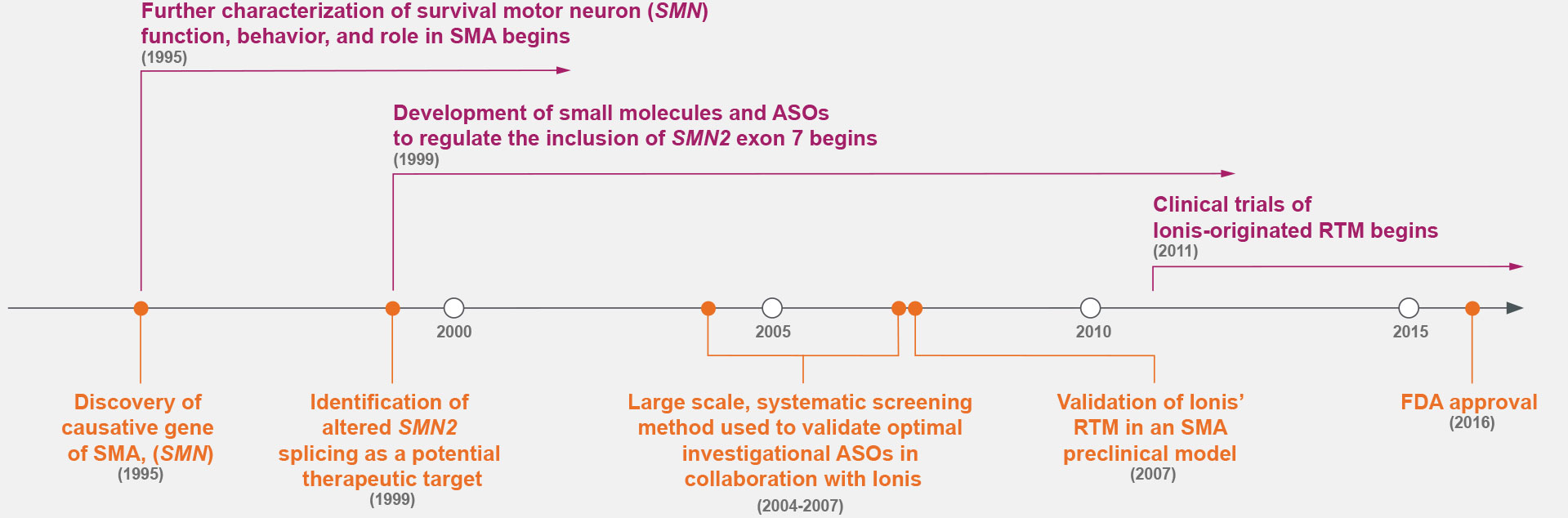
Case Study: SMA
Prior to the 1990s, the fundamental cause of SMA was unknown.7,14 Research throughout the 1990s identified the cause of SMA—the deletion of survival motor neuron gene 1 (SMN1)—and a potential therapeutic target: alternative splicing to include exon 7 of SMN2.7,9
ASOs were screened for the optimal target along exon 7 of SMN2, marking a shift in drug discovery.5,7-9,15
Ionis Has Decades of Experience Developing Therapies
for Hard-to-Treat Genetic Conditions3,5,7,9,15
Identifying the Neurologic Medicines of Tomorrow
Years of preclinical studies have yielded a rigorous process to optimize the identification and development of RNA-targeted medicines with reduced toxicity and a potentially favorable safety and tolerability profile for patients with neurologic diseases.1,3,16-19
Ionis is continually advancing the design, screening rigor, and development of its RNA-targeted medicines to optimize the selection of investigational medicines.1,3,18
Transforming Emerging Insights Into Investigational Medicines
Ionis Is Continually Advancing the Design, Screening, and Development of Its Investigational RNA-Targeted Medicines for Neurologic Diseases1,3,18,19
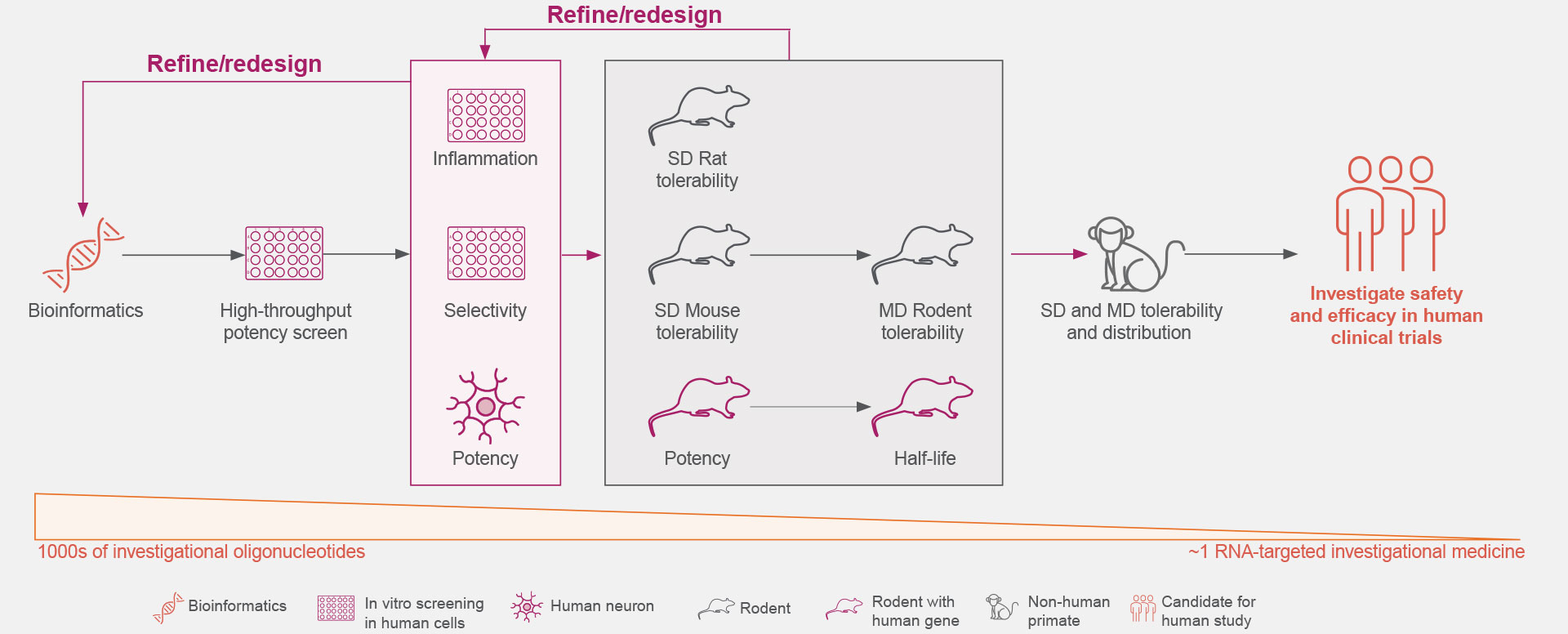
Click on the figure above to learn more about the rigorous screening process that allows the identification of an investigational RNA-targeted medicine from thousands of oligonucleotides.18,19
Ionis is continually refining and optimizing its RNA-targeted medicine platform to positively impact the lives of patients with neurologic diseases, creating new standards of care.24
Optimal genetic targets for Ionis’ investigational RNA-targeted medicines are identified preclinically to have the desired effect on the disease-associated protein.1,21
These RNA-targeted medicines target genes are then screened against our comprehensive genomic and experimentally trained databases to ensure they are genetically validated targets that play key roles in pathophysiological processes.1,21
Thousands of ASOs and hundreds of gene-specific binding sites are screened for optimal activity.1
The understanding of the molecular mechanisms of ASO-binding chemistry allows increased efficiency during the screening process.1
The Ionis iterative process extensively screens for increased potency and local inflammatory effects.1,18
Learn more about how chemical modifications may impact potency and inflammatory responses.1,18
The safety and tolerability of Ionis’ investigational RNA-targeted medicines are studied in preclinical disease models, including both rodent and non-human primates.1,18
These studies establish Ionis’ investigational RNA-targeted medicines’ ability to exhibit predictable kinetics, limited drug-drug interactions, and reduced off-target effects.5,22,23
Ionis’ broad, diverse pipeline has more than 30 agents with the potential to be ground-breaking therapies.24
Ionis has a wide range of investigational RNA-targeted medicines in clinical trials designed to evaluate safety and tolerability in patients with hard-to-treat neurologic diseases.24

